
SARS-CoV-2 infection proceeds through the binding of viral surface spike protein to the human ACE2 protein. Global spread of the infection has led to the evolution of the virus into more infective and transmissible variants with increased adaptation both in human and non-human hosts. Molecular simulations of the binding event between the spike protein and the ACE2 protein offer a route to assess potential increase or decrease in infectivity by measuring the change in strength of binding. We computationally contrasted the binding interactions between human ACE2 and coronavirus spike protein receptor binding domain (RBD) of the 2002 epidemic-causing SARS-CoV-1, SARS-CoV-2, and bat coronavirus RaTG13 using the Rosetta energy function. We find that the RBD of the spike protein of SARS-CoV-2 is highly optimized to achieve very strong binding with human ACE2 (hACE2) which is consistent with its enhanced infectivity. By contrasting the spike protein SARS-CoV-2 Rosetta binding energy with ACE2 of different livestock and pet species we find strongest binding with bat ACE2 followed by human, feline, equine, canine and finally chicken. This is consistent with the hypothesis that bats are the viral origin and reservoir species. These results offer a computational explanation for the increased infection susceptibility by SARS-CoV-2 and allude to therapeutic modalities by identifying and rank-ordering the ACE2 residues involved in binding with the virus.
Figure Caption: Schematic on the left is an illustration of the molecular events that occur during the first step of SARS-CoV-2 infection. The pictures on the right show a contrast in molecular interactions that occur at the interface of hACE2 protein when bound to SARS-CoV-1, SARS-CoV-2 and RaTG13
We further trained a neural network model, NN_MM-GBSA, that can accurately map simulated binding energies to experimental changes in binding strength upon amino acid changes in the viral RBD protein. The model utilizes binding energy components derived from MM-GBSA simulations performed on Molecular Dynamics trajectories of wild-type and mutant RBD proteins. This work can be valuable for predicting the infection potential of new SARS-CoV-2 variants. This can be a very valuable tool to predict host adaptation of the virus.

Figure Caption: Schematic representation of the workflow for building NN_MM-GBSA model. (A) MD simulations were performed for each single point amino acid substitution variant in explicit solvent followed by MM-GBSA analysis to calculate the decomposed components of binding energies. (B) MM-GBSA binding energy components were fed as inputs to the Neural network with the experimental Kd,app ratios as the regression target. The model is trained using five cycles of the five-fold cross validation procedure.
Insertions and deletions (indels) in protein sequences alter the residue spacing along the polypeptide backbone and consequently open up possibilities for tuning protein function in a way that is inaccessible by amino acid substitution alone. We describe an optimization-based computational protein redesign approach centered around predicting beneficial combinations of indels along with substitutions and also obtain putative substrate-docked structures for these protein variants. This modified algorithmic capability would be of interest for enzyme engineering and broadly inform other protein design tasks. We highlight this capability by (1) identifying active variants of a bacterial thioesterase enzyme (‘TesA) with experimental corroboration, (2) recapitulating existing active TEM-1 β-Lactamase sequences of different sizes, and (3) identifying shorter 4-Coumarate:CoA ligases with enhanced in vitro activities toward non-native substrates.
Figure Caption: The Steps of the IPRO+/− Design Cycle
We also developed a Python based open-source tool, Indel-Maker, that can be used to construct computational models of user-defined protein variants with specific indels and substitutions. This would be instrumental in discerning biophysical cues in experimentally tested variants to explain altered substrate/cofactor affinities or mutant stabilities.
Figure Caption: Indel-Maker requires users to provide an input file specifying insertions, deletions, and modifications to be performed on the input PDB. The required modifications are performed one by one, each followed by the loop closure (in the case of indels) and structure minimization using Rosetta's relax protocol (using the Rosetta all-atom force field). The resulting PDB and its corresponding Rosetta energy score are output at the end of each modification.
Monodispersed angstrom-size pores embedded in a suitable matrix are promising for highly selective membrane-based separations. They can provide substantial energy savings in water treatment and small molecule bioseparations. Such pores present as membrane proteins (chiefly aquaporin-based) are commonplace in biological membranes but difficult to implement in synthetic industrial membranes and have modest selectivity without tunable selectivity. Here we present PoreDesigner, a design workflow to redesign the robust beta-barrel Outer Membrane Protein F as a scaffold to access three specific pore designs that exclude solutes larger than sucrose (>360 Da), glucose (>180 Da), and salt (>58 Da) respectively. PoreDesigner also enables us to design any specified pore size (spanning 3–10 Å), engineer its pore profile, and chemistry. These redesigned pores may be ideal for conducting sub-nm aqueous separations with permeabilities exceeding those of classical biological water channels, aquaporins, by more than an order of magnitude at over 10 billion water molecules per channel per second.
Figure Caption: PoreDesigner for tuning solute selectivity in a robust and highly permeable outer membrane pore
Enzymes are highly specific catalysts that give rise to extraordinary rate enhancements under biological conditions. We developed a computational method called OptZyme that can be used to design enzymes when experimental techniques may be ineffective or infeasible. OptZyme employs the Iterative Protein Redesign & Optimization procedure (IPRO) to improve binding to substrates or to transition state analogues, which are used as proxies for the typically unknown transition state structure. Substrate binding was shown to correlate with the Michaelis constant and transition state analogue binding correlated with the enzyme’s turnover number. OptZyme has been applied to alter substrate specificity of E. coli beta-glucuronidase and acyl-ACP thioesterase, as well as Methanosarcina acetivorans methyl-coenzyme M reductase.
Figure Caption: Quaternary structure and catalyzed reaction for E. coli beta-glucuronidase.
Antibodies are proteins with important and significant medicinal and experimental applications. In this project, we are developing methods for the de novo design of antibodies. The Optimal Complementarity Determining Regions method (OptCDR) is a four-step procedure to design the binding pockets of antibodies. First, canonical structures that are likely to have favorable binding with the specified antigen are selected for the six complementarity determining regions (CDRs) of the antibody. This is followed by the initialization of the amino acid sequences of the CDRs using a rotamer library and an energy-minimizing MILP optimization formulation. Next, a modified version of our Iterative Protein Redesign & Optimization (IPRO) procedure is used to simultaneously modify the backbones and amino acids of the CDRs. Finally, the rotamer library and MILP optimization formulation are used again to generate a library of antibody binding pockets.

Figure Caption: Two sets of OptCDR designed CDRs binding distinct epitopes of vascular endothelial growth factor.
In this project, we aim to develop a new computational workflow utilizing highly accurate hybrid quantum mechanics/molecular mechanics (QM/MM) techniques for the design of enzymes. Specifically, mutants will be initially screened with the modified Iterative Protein Redesign and Optimization framework (IPRO) at the ground and transition states for improved binding energy of an alternative substrate (i.e. ethane) to a desired enzyme (i.e. Cytochrome P450 BM3). Next, mutants will be screened with potential energy surface scans along the reaction coordinate utilizing QM/MM through the Gaussian 03 framework. Successful designs screened by QM will next be experimentally constructed and tested, leading to an improved enzyme design for an alternative substrate. The P450 BM3 system was chosen to address the timely issue of controlled hydroxylation of small gaseous hydrocarbons (methane, ethane, and propane).
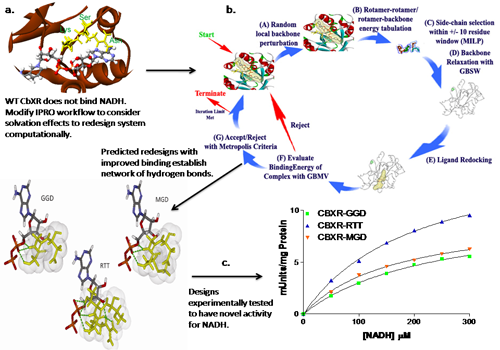
In this project, we computationally designed Candida boidinii xylose reductase (CbXR) to alter its cofactor specificity from NADPH to NADH. After compiling and comparing sequence information from previous studies involving cofactor switching mutations, we determined that their effect could not be explained as straightforward changes in volume, hydrophobicity, charge, or BLOSUM62 scores of the residues populating the cofactor binding site. Instead, we found that the use of a detailed cofactor binding energy function was needed to adequately capture the relative affinity towards different cofactors. The implicit solvation models Generalized Born with molecular volume integration and Generalized Born with simple switching were integrated in the Iterative Protein Redesign and Optimization (IPRO) framework to drive the redesign of CbXR to function using the non-native cofactor NADH. We identified ten variants that improve the calculated interaction energy with NADH by introducing mutations in the CbXR binding pocket. These protein variants were experimentally tested, and seven out of ten possessed xylose reductase activity utilizing NADH while essentially abolishing NADPH-dependent activity. Given the higher stability of NADH relative to NADPH, and the higher cost of NADPH regeneration compared to NADH generation, a NADH-utilizing CbXR variant may prove industrially useful. More importantly, this method can be extended to design other enzyme-cofactor systems to utilize more stable and more abundant cofactors.
In this research, we use atomistic potential energy functions to design small numbers of protein structures with novel or modified functions. One of the many challenging tasks of protein design is the introduction of a completely new function into an existing protein scaffold. We have developed the OptGraft procedure for placing a novel binding pocket onto a protein structure so as its geometry is minimally perturbed. This is accomplished through a two-level procedure where we first identify where are the most appropriate locations to graft the new binding pocket into the protein fold by minimizing the departure from a set of geometric restraints using mixed-integer linear optimization. On identifying the suitable locations that can accommodate the new binding pocket, CHARMM energy calculations are employed to identify what mutations in the neighboring residues, if any, are needed to ensure that the minimum energy conformation of the binding pocket conserves the desired geometry. This computational framework was benchmarked against the results available in the literature for engineering a copper binding site into thioredoxin protein. OptGraft has been used to guide the transfer of a calcium-binding pocket from thermitase protein (PDB: 1thm) into the first domain of CD2 protein (PDB: 1hng). Experimental characterization of three de novo redesigned proteins with grafted calcium-binding centers demonstrated that they all exhibit high affinites for terbium and can selectively bind calcium over magnesium.
Figure Caption: A schematic presentation of OptGraft. Step 1 identifies where the binding pocket should be grafted on the acceptor protein. Step 2 determines what residue modifications are required in the surrounding region (shown in yellow) to ensure that the correct binding pocket geometry is preserved.
We have also worked with researchers from the Department of Biological Sciences at the Korea Advanced Institute of Science and Technology to rationally design the substrate specificity of D-hydantoinase because enzymes that exhibit superior catalytic activity, stability and substrate specificity are highly desirable for industrial applications. These goals prompted the designed substrate specificity of Bacillus stearothermophilus D-hydantoinase toward the target substrate hydroxyphenylhydantoin. Positions crucial to substrate specificity were selected using structural and mechanistic information on the structural loops at the active site. The size and hydrophobicity of the involved amino acids were rationally changed, and the substrate specificities of the designed D-Hyd mutants were investigated. As a result, M63I/F159S exhibited about 200-fold higher specificity than the wild-type enzyme. Systematic mutational analysis and computational modeling also supported the rationale used in the design.
In this research, we developed the computational procedure IPRO (Iterative Protein Redesign and Optimization procedure) for the redesign of an entire combinatorial protein library in one step using energy based scoring functions. IPRO relies on identifying mutations in the parental sequences that, when propagated downstream in the combinatorial library, improve the average quality of the library (e.g., stability, binding affinity, specific activity, etc.). Residue and rotamer design choices are driven by a globally convergent Mixed-Integer Linear Programming (MILP) formulation. Unlike many of the available computational approaches, the procedure allows for backbone movement as well as re-docking of the associated ligands after a pre-specified number of design iterations. IPRO can also be used, as a limiting case, for the redesign of a single or handful of individual sequences. The application of IPRO was highlighted through the redesign of a sixteen member library of E. coli/B. subtilis dihydrofolate reductase hybrids, both individually and through upstream parental sequence redesign, for improving the average binding energy. Computational results demonstrate that it is indeed feasible to improve the overall library quality as exemplified by binding energy scores through targeted mutations in the parental sequences.
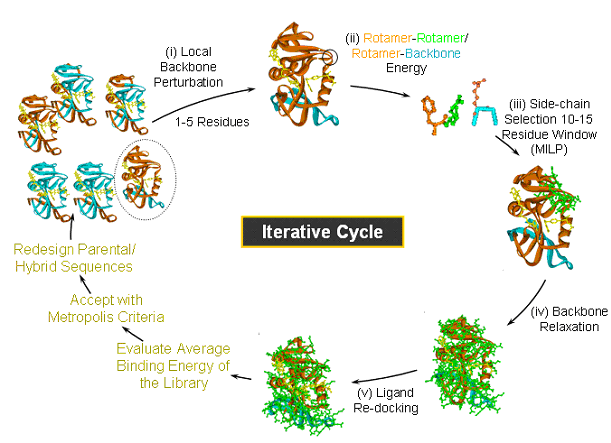
Figure Caption: IPRO is an iterative protein redesign procedure that includes the following steps: (i) A local region of the protein (1-5 consecutive residues as shown in black circle) is randomly selected for perturbation. The backbone torsion angles of these residues are perturbed by up to ±5 degrees. (ii) All amino acid rotamers consistent with these torsion angles are selected at each position from the Dunbrack and Cohen rotamer library.. Rotamer-backbone and rotamer-rotamer energies are calculated for all the selected rotamers using a suitable energy function (e.g., CHARMM). (iii) A mixed-integer linear programming formulation is used to select the optimal rotamer at each of these positions such that the binding energy is minimized. (iv) The backbone of the protein is relaxed through energy minimization in order to allow it to adjust to these new side-chains. (v) The ligand position is re-adjusted with respect to the modified backbone and side-chains using the ZDOCK docking software. (vi) The binding energy of the protein-ligand complex is evaluated and the move is accepted or rejected using the Metropolis criterion.
We have extended the IPRO framework for the design of protein libraries with a targeted ligand specificity. Mutations that minimize the binding energy with the desired ligand are identified. At the same time, explicit constraints are introduced that maintain the binding energy for all decoy ligands above a threshold necessary for successful binding. The modified framework was demonstrated by computationally altering the effector binding specificity of the bacterial transcriptional regulatory protein AraC, belonging to the AraC/XylS family of transcriptional regulators for different unnatural ligands. The obtained results demonstrate the importance of systematically suppressing the binding energy for competing ligands. By pinpointing a small set of mutations within the binding pocket, the difference in binding energies between targeted and decoy ligands, even when very similar, is maximized.
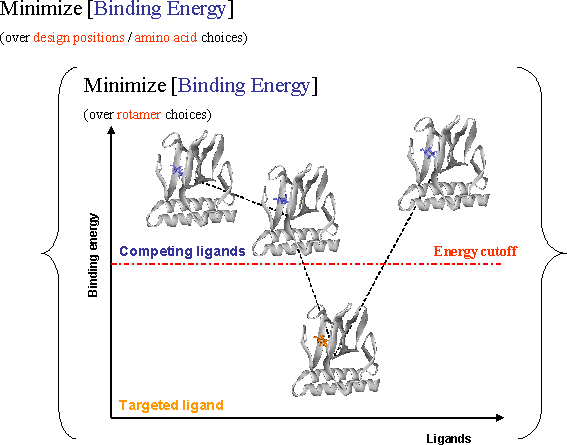
Figure Caption: Modified IRPO as a two-level optimization problem. In the outer level new designs with respect to amino acids choices are generated, while the inner level makes the rotamer choices that minimize the binding energy with respect to various ligands. The amino acids are chosen such that the binding energy with respect to all undesired ligands is above a cutoff value preventing their binding while simultaneously ensuring sufficiently low binding energy with respect to the targeted ligand.
In this research we have introduced a computational procedure, OPTCOMB (Optimal Pattern of Tiling for COMBinatorial library design), for designing protein hybrid libraries that optimally balance library size with quality. The proposed procedure is directly applicable to oligonucleotide ligation-based protocols such as GeneReassembly, DHR, SISDC, and many more. Given a set of parental sequences and the size ranges of the parental sequence fragments, OPTCOMB determines the optimal junction points (i.e., crossover positions) and the fragment contributing parental sequences at each one of the junction points. By rationally selecting the junction points and the contributing parental sequences, the number of clashes (i.e., unfavorable interactions) or any other scoring metric in the library is systematically optimized with the aim of improving the overall library quality.
Clashes are identified by characterizing all contacting residue pairs present in protein hybrids for inconsistency with protein family structural features. This approach is based on examining contacting residue pairs with different parental origins for different types of potentially unfavorable interactions (i.e., electrostatic repulsion, steric hindrance, cavity formation and hydrogen bond disruption). The identified clashing residue pairs between members of a protein family are then contrasted against functionally characterized hybrid libraries (FamClash Procedure).
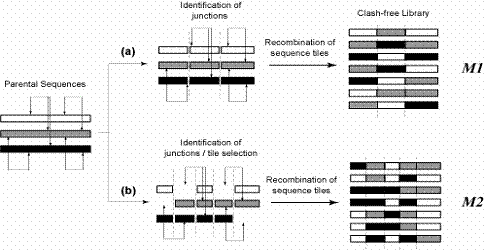
Figure Caption: A pictorial representation of an example where three parental sequences form a combinatorial library through recombination. The clashes between different residues are shown as double-headed arrows. The junction points are shown as dashed lines. The combinatorial libraries are designed using two different design rules: (a) all parental sequences contribute fragments at each of the junction points (Model M1), and (b) selective restrictions are imposed on the set of oligomers being contributed by the parents (Model M2).
We have also developed the S2 scoring system, which uses amino acid property bins to identify clashes in protein hybrids and also quantify the degree of their mismatched interactions. A cytochrome P450 library (Otey et al (2006)) was used for benchmarking, and the S2 scoring system was found to be able to significantly functionally enrich the library compared with other scoring systems. Given this scoring base, we implemented S2 in OPTCOMB and also developed OPTOLIGO, a formulation for optimally designing protein combinatorial libraries involving mutations. Computational benchmarking results demonstrate the efficacy of OPTCOMB and OPTOLIGO to generate high-scoring libraries of a pre-specified size.

Figure Caption: The 20 amino acids were divided into bins for the properties of volume, charge, and hydrophobicity. The bins were created so that the maximum difference between the property values of two amino acids in a given bin was less than the minimum difference between an amino acid in that bin and one from an adjacent bin.
[Pantazes et al (2007); Saraf et al (2005); Saraf et al (2004); Moore and Maranas (2004); Saraf and Maranas (2003); Moore and Maranas (2003); Saraf et al (2003)]
Work in our group examined for the first time how fragmentation length, annealing temperature, sequence identity and number of shuffled parental sequences affect the number, type and distribution of crossovers along the length of full-length reassembled sequences. In the eShuffle framework, annealing events during reassembly are modeled as a network of reactions, and equilibrium thermodynamics along with complete nucleotide sequence information was employed to quantify their conversions and selectivities. Comparisons of eShuffle predictions against experimental data revealed good agreement, particularly in light of the fact that there were no adjustable parameters. Specifically we found that crossover numbers were boosted by reducing fragmentation length and annealing temperature and that crossovers tend to aggregate in regions of near perfect sequence identity.
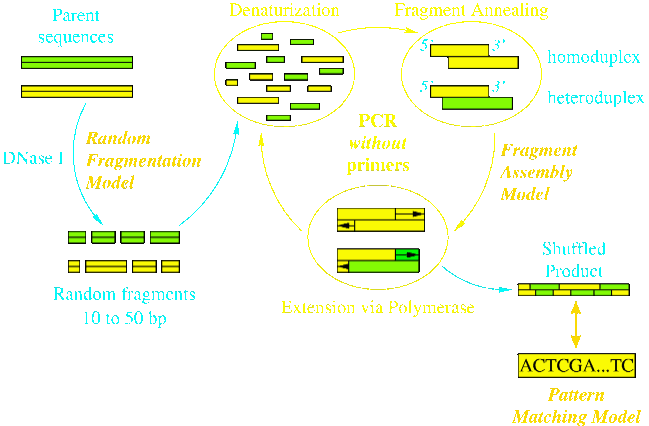
Figure Caption: DNA Shuffling Protocol
The customization of eShuffle for the SCRATCHY protocol led to the eSCRATCHY framework. Using eSCRATCHY we found that in SCRATCHY libraries (i) fragmentation length used for reassembly does not influence the number or location of crossovers generated in full-length sequences, (ii) the crossover distribution is shaped by the crossover statistics of the ITCHY library, and (iii) crossovers are spread evenly throughout the crossover region. The need to safeguard against the formation of reassembled sequences with either truncated or duplicated domains motivated us to further extend the eShuffle framework to consider out-of-sequence annealing event. Instead of “locking” fragments into their alignment positions, the annealing free energy change was used to determine the likelihood of duplex formation, allowing the prediction of the relative frequency that fragments from different sequence regions will anneal during reassembly.
We have also explored the possibility of boosting or even specifically redirecting the formation of crossovers in DNA shuffling by exploiting the inherent redundancy in the codon representation (e.g., isoleucine has the following three synonymous codon representations: ATA, ATC and ATT) while complying with host preferences for specific patterns of codon usage.
[Moore and Maranas (2003b); Moore and Maranas (2002b); Moore and Maranas (2002a); Lutz et al (2001); Moore et al (2001); Moore and Maranas (2000); Moore et al (2000)]